

01
Description
More about
02
Short list of features
More about
03
Technical Description
More about
04
Repositories and Download
More about
YAMBO is an open-source code released within the GPL licence that implements ground-state as well as excited-state properties in an ab initio context. The code implements MBPT, DFT and Non-Equilibrium Green's Function Theory (NEGF) in order to allow the user to calculate a wealth of physical properties: reliable band gaps, band alignments, defect quasi-particle energies, optical and non--equilibrium properties. YAMBO resorts to previously computed electronic structure, and for this reason it is interfaced with other DFT codes, among which Quantum ESPRESSO.
Among the variety of physical quantities that can be described with YAMBO, we mention:
- Electronic properties: quasi-particle energies, line-widths, and renormalization factors;
- Linear optical properties, capturing the physics of excitons, plasmons, and magnons;
- Temperature dependent electronic and optical properties via electron-phonon coupling;
- Non--equilibrium and Non--linear optical properties via NEGF real--time simulations;
- Advanced post-processing tools to analyse the simulation flow of data.
All these properties are ubiquitous for the understanding of the optical and electronic properties of a wealth of advanced materials. More importantly, YAMBO provides a unique approach for the non--equilibrium regime, where ab initio numerical tools are scarse, able to model Pump\&Probe experiments and to capture coherent electron dynamics. The code is under a constant development and fully documented.
- Diffusion
- Performance in HPC enviroments
Diffusion
YAMBO has attracted over the years a growing community of code users and developers. The code is routinely used to organise hands--on schools where the most fundamental concepts of the underling theory are described. A dedicated user forum (with more than 900 subscribers) is actively used to answer users' questions and doubts. The code has been used to produce the results published in hundreds of papers by many different groups all over the world. The YAMBO reference papers [1,2] have been cited more than 800 times to date (Mar 2022). At the moment YAMBO counts about 20 active developers and the source project is publicly hosted on the github platform.
Performance in HPC environments
YAMBO has a user-friendly command-line interface, flexible I/O procedures, and it is parallelised by using an hybrid MPI plus OpenMP infrastructure, very well integrated with support of GPGPU-based heterogeneous architectures. This makes it possible to distribute the workload to a large number of parallel levels. In practice, depending on the kind of calculation, all the variables to be used (k/q grids, bands, quasi-particles, etc) are distributed along the different level of parallelisation. At present YAMBO has been shown to be efficient in large-scale simulations (several upto few tens of thousands MPI tasks combined with OpenMP parallelism) for most of its calculation environments.
The GPU porting was first made using CUDA-Fortran, and more recently enlarged to other programming models (like OpenACC and OpenMP5, both in development). To avoid code duplication, we make an intense use of pre-processor macros that activate the language chosen at compile time. This allows YAMBO to optimally integrate MPI-OpenMP with programming models for GPGPU. The outcome of this integration is well exemplified by the scaling tests reported in figure, for the calculation of quasi-particle corrections on a graphene/Co interface (GrCo) composed by a graphene sheet adsorbed on a Co slab 4 layers thick, and a vacuum layer as large as the Co slab.
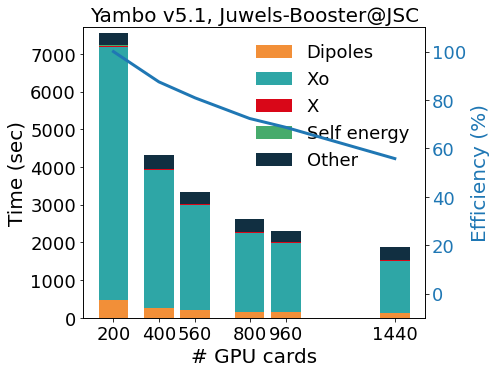
The test represents a prototype calculation, as it involves the evaluation of a response function, of the Hartree-Fock self-energy and, finally, of the correlation part of the self-energy. The scalability and relative efficiency are reported in the Figure as a function of the number of GPU cards and show a very good scalability up to 1440 GPUs (360 nodes on Juwels-Booster@JSC, 4 NVIDIA A100 per node).
References:
-
[1] D. Sangalli et al., Many-body perturbation theory calculations using the yambo code, J. Phys.: Condens. Matter 31, 325902 (2019).
-
[2] A. Marini, M. Gruning, D. Varsano and C. Hogan, Yambo: An ab initio tool for excited state calculations, Comp. Phys. Comm. 180, 1392 (2009).
YAMBO is a plane-wave ab-initio code for calculating quasiparticle energies and optical properties of electronic systems within the framework of many-body perturbation theory and time-dependent density functional theory. Quasiparticle energies are calculated within the GW approximation for the self-energy. Optical properties are evaluated either by solving the Bethe–Salpeter equation or by using the adiabatic local density approximation. With YAMBO you can perform:
- GW calculations for electronic excitation within the plasmon-pole approximation or using full-frequency GW.
- Calculations of electron loss and optical absorption spectra of solids, and dynamical polarizability of molecules at different levels of theory.
- Calculation of excitons and excitonic properties of solids evaluated soling the Bethe-Salpether equation.
- Study of electron-hole excitations in the presence of spin-orbit coupling. Spin–orbit coupling and Kerr effect within a fully noncollinear BSE framework.
- Time dependent DFT calculations.
- Electron-phonon and excitation-phonon interaction, influence of the temperature on the electronic structure and optical spectra.
- Real-time propagation of the density matrix and Bloch states for nonlinear optics.
Software
YAMBO is a FORTRAN/C code used worldwide to calculate quasiparticle corrections and excitonic properties of materials by using the GW+ BSE method. YAMBO relies on the KS data generated by QE. The code is parallelized over several MPI+OpenMP levels. YAMBO stores information in several database files (few tens), the biggest reaching few GBs in size for our systems, for which a NetCDF/HDF5 format is adopted, optimizing IO and data portability. The code has been extensively tested and used on different HPC architectures, for large scale systems. Recent developments in YAMBO concern the implementation of a new algorithm to reduce the number of empty states needed to converge GW calculations (X and G terminators), the implementation of dense parallel linear algebra to make diagonalisation and inversion of large matrices more efficient, and the resolution of memory and parallelism bottlenecks, according to the results of performance profiling. The YAMBO implementation of GW is parallel on the k/q grids, bands summation and quasiparticle energies, using a hybrid MPI-OpenMP approach. Explicit OpenMP support is implemented following different strategies according to the specific kernels. The BSE routines are parallel on k-points, electron-hole basis elements, and transitions. GW and BSE calculations are computationally expensive and, for complex materials and surfaces, can be performed only exploiting the resources offered by modern Tier0 systems.
Particular Libraries
YAMBO is an open source codes distributed under GNU General Public Licence. The code source is hosted on the Github hosting service https://github.com/yambo-code. YAMBO is a Fortran/C code that exploits a number of optimized libraries such as BLAS, LAPACK, FFTW, SCALAPACK, NetCDF/HDF5, PETSC, and SLEPC.
Parallel programming
YAMBO parallelism is based on hybrid MPI plus share-memory OpenMP strategy. YAMBO tends to use MPI parallelism as much as memory allows, then resorting to OpenMP parallelism. YAMBO makes use of several levels of MPI parallelism [S. Sangalli et al. J. Phys.: Condens. Matter 31 (2019) 325902]. The OpenMP multi-thread parallelization has been recently included at the low level when dealing with plane-wave summations and FFT.
I/O requirements
Electronic structure data, like for instance the wavefunctions, are written and read by QE’s PWscf executable using a direct access to file: each task uses a raw format to store its information. These files can be used to produce checkpoints (restart). YAMBO calculates quasiparticle corrections and solves the Bethe-Salpeter equation, reading and elaborating raw files generated by PWscf and managing the I/O using the NetCDF/HDF5 libraries. One of the most useful features of YAMBO is its ability to carry out the entire database I/O using the NetCDF/HDF5 libraries. YAMBO stores information in several database files (few tens), the biggest reaching few GBs in size for our systems. YAMBO relies on the KS wavefunctions generated by the DFT PWscf code through the interface p2y (PWscf to yambo interface). p2y can read and process band structure output generated by PWscf. Job-dependent database are also created at run-time, with information specific to each runlevel. Output files are created at the end of each calculation and are intended to human use (for instance for post-processing operations or for plotting). The creation of each database is controlled by its particular Fortran subroutine however, data writing is performed at a low level by common modules, in order to minimize problems associated with portability. YAMBO calculations can be restarted, making these codes suitable for HPC machines with very diverse scheduler policies. Relatively short wall time with large machine partitions can be safely exploited.